Overview
Overview of geneRNIB
geneRNIB is a cloud-hosted platform designed to evaluate gene regulatory network (GRN) inference methods in a standardized and reproducible way. It brings together datasets, GRN models, evaluation metrics, and a dynamic leaderboard to track the latest advancements in GRN benchmarks.
To ensure fair comparisons, geneRNIB provides five benchmark datasets, each tailored to assess different aspects of GRN inference. These datasets originate from the same cell types and experiments, allowing context-specific evaluations. The platform supports both transcriptomics-based GRN inference, which relies solely on gene expression data, and multi-omics approaches that integrate chromatin accessibility and gene expression to uncover regulatory interactions.
Evaluating GRN performance is challenging due to the lack of a definitive “ground truth” network. To address this, geneRNIB employs eight standardized evaluation metrics that use perturbation data to assess inferred interactions. The Wasserstein (WS) distance measures shifts in gene expression after perturbations, helping to determine how well a model captures true regulatory effects. Additionally, regression-based metrics (R₁ and R₂) assess predictive accuracy, ensuring that inferred regulatory links contribute to meaningful predictions.
To put GRN models into context, geneRNIB also includes three control models. A simple baseline computes Pearson correlations between genes, serving as a quick reference for benchmarking. A positive control model sets an upper bound by incorporating all available variation, while a negative control model generates random networks to ensure meaningful performance comparisons.
Built with modern computational tools like Docker and Viash, geneRNIB prioritizes scalability and reproducibility. It provides a structured framework for integrating new datasets, inference methods, and evaluation metrics, making it a powerful resource for advancing GRN research.
Installation
For installation, follow the task_gen_benchmark.
Once the repository is cloned and the required software installed, proceed to the next steps.
Download resources for GRN inference and evalation:
cd task_grn_benchmark
# download resources
scripts/download_resources.sh
The full resources is acceesible
Infer a GRN
viash run src/methods/dummy/config.vsh.yaml -- --multiomics_rna resources/grn-benchmark/multiomics_rna.h5ad --multiomics_atac resources/grn-benchmark/multiomics_atac.h5ad --prediction output/dummy.csv
Similarly, run the command for other methods.
Evaluate a GRN
scripts/run_evaluation.sh --grn resources/grn-benchmark/grn_models/collectri.csv
Similarly, run the command for other GRN models.
See examples of interacting with the framework can be found in section Examples.
—- TODO: improve this with info given above
The pipeline can evaluate algorithms that leverage only one of the multi-omic data types (RNA-Seq or ATAC-Seq) or both. It also evaluates the performance of two controls:
As a negative control, the pipeline evaluates the performance of a random network.
As a positive control, the pipeline evaluates the performance of a network derived from correlation of genes in the perturbation dataset used for evaluation.
The two types of regression models are:
Regression from GRN regulations to target expression
Regression from TF expression of predicted regulators to target expression
The evaluation is done with the help of pertubation data, using two different approaches:
Regression from GRN regulations to target expression
Regression from TF expression of predicted regulators to target expression
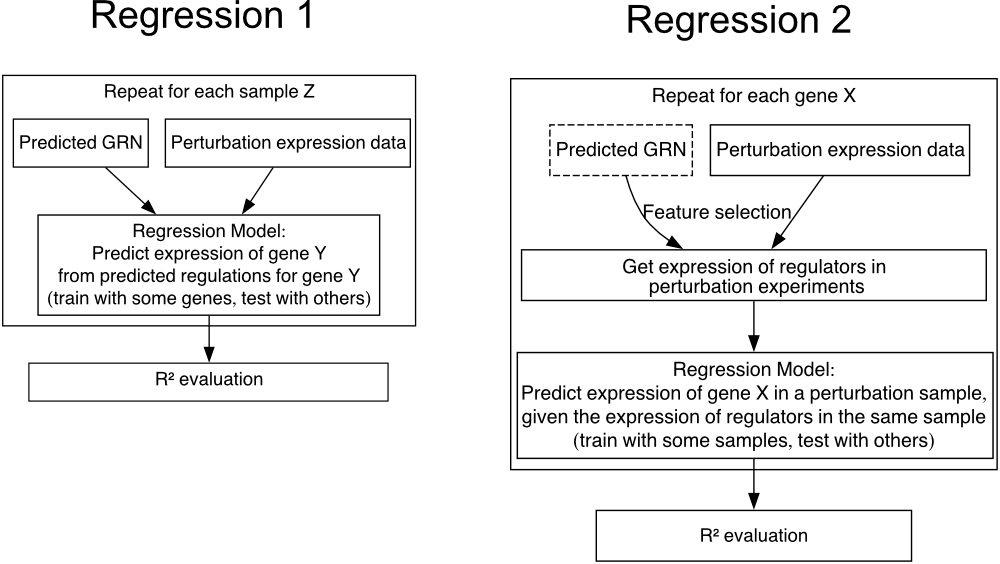
Evaluation 1: Regression from GRN regulations to target expression
The first approach we used is similar to GRaNPA and the multivariate decision tree in Decoupler, where regulatory weights from the GRN form the feature space to predict perturbation data. In this method, we train one model per sample. The feature space matrix has dimensions of genes by transcription factors (TFs), with values being the regulatory weights from the GRN or 0 if the link is absent. The target space matrix represents the perturbation data for each sample. We evaluate the model’s predictive performance using a 5-fold cross-validation scheme and the coefficient of determination (R²) as the metric. LightGBM is used for computational efficiency.
Evaluation 2: Regression from TF expression of predicted regulators to target expression
In the second approach, instead of using regulatory weights, we utilized the expression of putative regulators (TFs) from the perturbation data to construct the feature space. We fit one model per gene, selecting regulators based on the regulatory weights suggested by the GRNs. This method is similar to many modern GRN inference techniques.